NiPorph • NitroPy Rotationsbarrieren: Unterschied zwischen den Versionen
Zur Navigation springen
Zur Suche springen
| (13 dazwischenliegende Versionen desselben Benutzers werden nicht angezeigt) | |||
| Zeile 1: | Zeile 1: | ||
{{:NiPorph • Ligand Rotationsbarrieren}} | {{:NiPorph • Ligand Rotationsbarrieren}} | ||
== | == R=H: triplett-Rotationsbarriere als Energiedifferenz der 0° und 45° 1:1-t-Komplexe == | ||
die Hybridfunktionale wurden als singlepoint Rechnung auf die mit Dispersionsterm (blau) und | |||
ohne Dispersionsterm (rot) voroptimierten Strukturen (PBE/DZP und PBE/TZVP) aufgesetzt. | |||
Fazit: Fürs paper nur die auf PBE volloptimierten Barrieren. | |||
[[Datei:Rotationsbarriere Nitropyridin 1 zu 1 verschiedenen Dichte- und Hybridfunktionale.png | 800px ]] | |||
== Rotation um die R-NiPorph · NitroPy Bindung == | |||
{| {{table}} | {| {{table}} | ||
| Zeile 21: | Zeile 28: | ||
| 2t(coplanar-fern)||-||2.19 | | 2t(coplanar-fern)||-||2.19 | ||
|} | |} | ||
== Graphen 1:1 Komplexe == | |||
<gallery caption=1:1-Komplexe perrow=2 widths=600px heights=400px> | |||
File:Plot der H-NiPorph-NitroPy-Rotationsenergie PBE-DZP 1-s.png|1:1 singlett R=H | |||
File:Plot der H-NiPorph-NitroPy-Rotationsenergie PBE-DZP 1-t.png|1:1 triplett R=H | |||
File:Plot der FlourPhenyl-NiPorph-NitroPy-Rotationsenergie PBE-DZP 1-s.png|1:1 singlett R=FlourPhenyl | |||
File:Plot der FlourPhenyl-NiPorph-NitroPy-Rotationsenergie PBE-DZP 1-t.png|1:1 triplett R=FlourPhenyl | |||
</gallery> | |||
== Graphen 2:1 Komplexe == | |||
Die 2:1 Komplexe mit R=Flourphenyl sind asymmetrisch. | |||
Einer der Liganden ist näher ans Nickel gebunden, der andere bildet | |||
stärkere Wasserstoffbrücken mit den Flour-Atomen des Flourphenols. | |||
Im folgenden ist die Rotationdes näher gebundenen als 2:1 (coplanar nah), die | |||
des entfernter gebundenen als 2:1 (coplanar fern) bezeichnet. | |||
<gallery caption=2:1-Komplexe perrow=2 widths=600px heights=400px> | |||
File:Plot der H-NiPorph-NitroPy-Rotationsenergie PBE-DZP 2-s-coplanar.png|2:1 singlett R=H | |||
File:Plot der H-NiPorph-NitroPy-Rotationsenergie PBE-DZP 2-t-coplanar.png|2:1 triplett R=H | |||
File:Plot der FlourPhenyl-NiPorph-NitroPy-Rotationsenergie PBE-DZP 2-t-coplanar-fern.png|2:1 (coplanar fern) triplett R=Flourphenyl | |||
File:Plot der FlourPhenyl-NiPorph-NitroPy-Rotationsenergie PBE-DZP 2-t-coplanar-nah.png|2:1 (coplanar nah) triplett R=Flourphenyl | |||
</gallery> | |||
Aktuelle Version vom 11. Mai 2010, 15:26 Uhr
Rotation um den Winkel Phi von 0..90°

- Theoretische Untersuchungen zu Ni-Porphyrin • Ligand Komplexen
- NiPorph • Ligand Rotationsbarrieren
- NiPorph • Py Rotationsbarrieren
- NiPorph • NitroPy Rotationsbarrieren
R=H: triplett-Rotationsbarriere als Energiedifferenz der 0° und 45° 1:1-t-Komplexe
die Hybridfunktionale wurden als singlepoint Rechnung auf die mit Dispersionsterm (blau) und ohne Dispersionsterm (rot) voroptimierten Strukturen (PBE/DZP und PBE/TZVP) aufgesetzt.
Fazit: Fürs paper nur die auf PBE volloptimierten Barrieren.

Rotation um die R-NiPorph · NitroPy Bindung
| R=H | R=FlourPhenyl | |
| 1s | 0.20 | 1.03 |
| 1t | 1.21 | 1.88 |
| 2t(d2d) | 0.85 | - |
| 2t(coplanar-nah) | - | 1.84 |
| 2t(coplanar-fern) | - | 2.19 |
Graphen 1:1 Komplexe
- 1:1-Komplexe
-
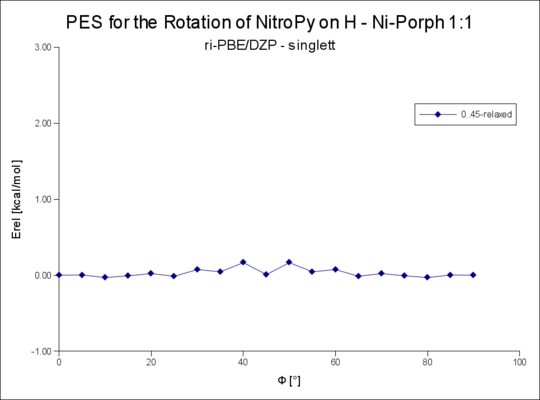 1:1 singlett R=H
1:1 singlett R=H -
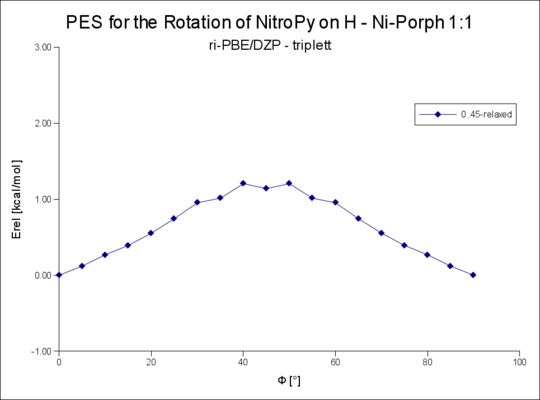 1:1 triplett R=H
1:1 triplett R=H -
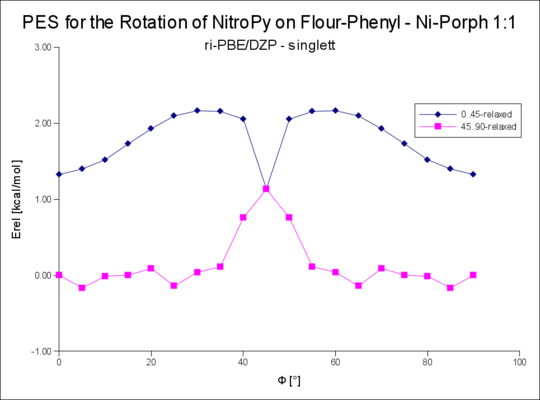 1:1 singlett R=FlourPhenyl
1:1 singlett R=FlourPhenyl -
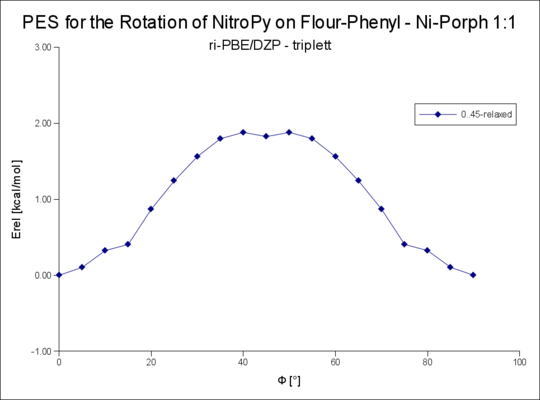 1:1 triplett R=FlourPhenyl
1:1 triplett R=FlourPhenyl




Graphen 2:1 Komplexe
Die 2:1 Komplexe mit R=Flourphenyl sind asymmetrisch. Einer der Liganden ist näher ans Nickel gebunden, der andere bildet stärkere Wasserstoffbrücken mit den Flour-Atomen des Flourphenols.
Im folgenden ist die Rotationdes näher gebundenen als 2:1 (coplanar nah), die des entfernter gebundenen als 2:1 (coplanar fern) bezeichnet.
- 2:1-Komplexe
-
2:1 singlett R=H
-
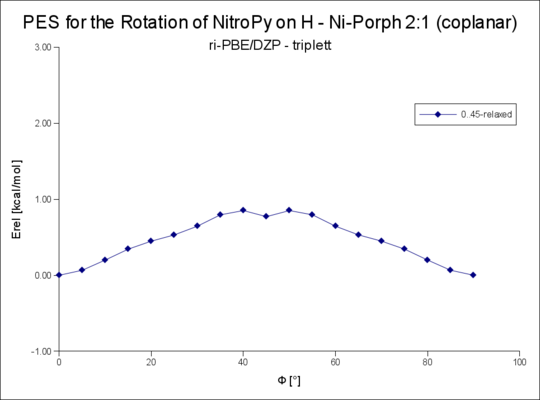 2:1 triplett R=H
2:1 triplett R=H -
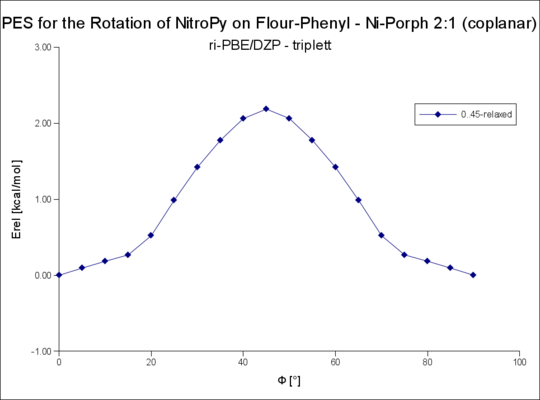 2:1 (coplanar fern) triplett R=Flourphenyl
2:1 (coplanar fern) triplett R=Flourphenyl -
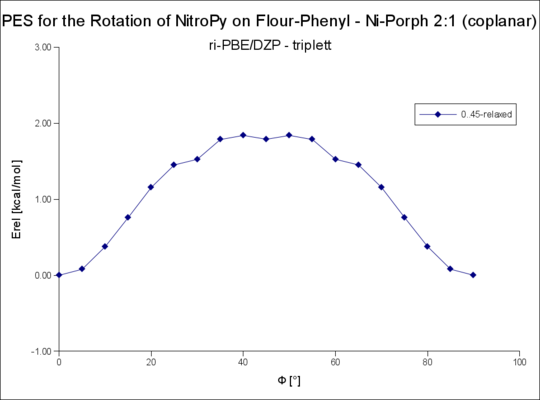 2:1 (coplanar nah) triplett R=Flourphenyl
2:1 (coplanar nah) triplett R=Flourphenyl



{kind=link}